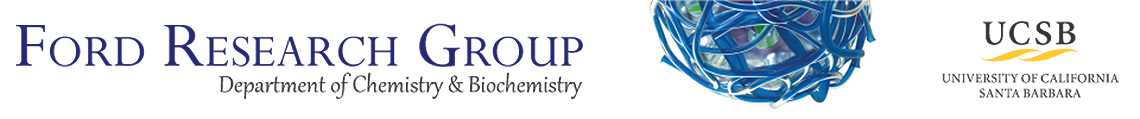
Recent & Highlighted Publications & Presentations

Papers by subject
Photochemistry/Photophysics
Bioinorganic Chemistry of SMBs
Sustainable Chemistry & Biomass Conversion
Presentations by subject
Biomass/Sustainable Chemistry
Photochemistry/Small Molecule Bioregulators
Photochemistry/Photophysics Papers
Optical oxygen sensing by MPA-capped CdTe quantum dots immobilized in mesoporous silica
L. P. Ravaro, P. C. Ford, A. S. S. de Camargo.
Microporous and Mesoporous Materials, 2020, 303, 110237
DOI: 10.1016/j.micromeso.2020.110237
Abstract
A novel nanocomposite luminescent material was prepared by taking advantage of the versatile wet impregnation method for the dispersion of CdTe quantum dots (QDs) into mesoporous silica host matrix and thus providing great interaction between oxygen and QDs, with potential application in an optical oxygen sensor. The optical/spectroscopic properties of the QDs suspended in aqueous media and incorporated in mesoporous silica were evaluated as a function of aging time, tempera-ture variation and oxygen concentration. Luminescence quenching studies were carried out for both QDs suspended in solution and loaded into the silica matrix, in the presence of varying O2concentration. By Stern-Volmer plot analysis, obtained at different temperatures, it was possible to verify the existence of two types of emission quenching mechanisms for CdTe QDs. After aging for 120 days at room temperature, the QDs in colloidal suspension displayed a small red-shifted emission, which was interpreted as a decreased bandgap energy owing to the increase in the nano-crystal size. In contrast, the emission spectrum of CdTe QDs loaded into the mesoporous SiO2 matrix remained unchanged after aging for the same time at ambient temperature. The present-ed results will contribute to the discernment of oxygen quenching mechanisms and chemical stabil-ity of optical sensors based on CdTe QDs.
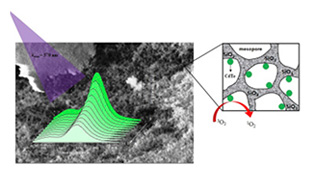
Near-Infrared and Visible Photoactivation to Uncage Carbon Monoxide from an Aqueous-Soluble PhotoCORM
Q. Jiang, Y. Xia, J. Barrett, A. Mikhailovsky, G. Wu, D. Wang, P. Shi, P. C. Ford.
Inorganic Chemistry, 2019, 58, 11066-11075
DOI: 10.1021/acs.inorgchem.9b01581
Abstract
Multiphoton excitation allows one to access high energy excited states and perform valuable tasks in biological systems using tissue penetrating near-infrared (NIR) light. Here, we de-scribe new photoactive manganese tricarbonyl complexes incorporating the ligand 4′-p-N,N-bis(2-hydroxyethyl)amino-benzyl-2,2′:6′,2″-terpyridine (TPYOH), which can serve as an an-tenna for two photon NIR excitation. Solutions of Mn(CO)3(TPYOH)X (X = Br– or CF3SO3–) complexes are very photoactive toward CO release under visible light excitation (405 nm, 451 nm). The same responses were also triggered by multiphoton excitation at 750 and 800 nm. In this context, we discuss the potential applications of these complexes as visible/NIR light photoactivated carbon monoxide releasing moieties (photoCORMs). We also report the isola-tion and crystal structures of the TPYOH complexes Mn(TPYOH)Cl2 and [Mn(TPYOH)2](CF3SO3)2, to illustrate a possible photolysis product(s).
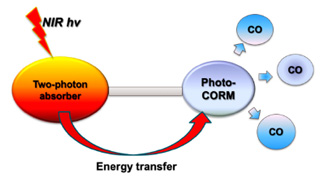
Nitric Oxide Uncaging from a Hydrophobic Chromium(III) PhotoNORM: Visible and Near-Infrared Photochemistry in Biocompatible Polymer Disks
P.-J. Huang, J.V. Garcia, A. Fenwick, G. Wu, P. C. Ford
ACS Omega 2019, 4, 9181−9187
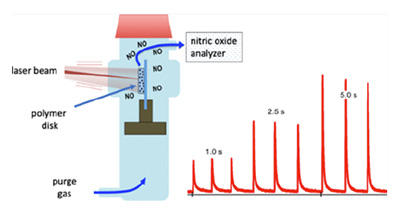
Abstract
Devices consisting of polymer disks (PDs) of optically clear or translucent, medical-grade silicone loaded with a new hydrophobic, oxygen-stable, photoactivated nitric oxide-releasing moiety (pho-toNORM) are described. The photoNORM is the new O-nitrito chromium(III) complex trans-[Cr(PetA)(ONO)2](BF4) (PetA = 5,14-dimethyl-7,12-diphenyl-1,4,8,11-tetraaza-cyclotetradecane), of which the synthesis, X-ray crystal structure, and solution-phase photochemistry are described. Several different commercially available silicone polymers were tested with this photoNORM, and nitric oxide photo-uncaging with 451 nm light from these systems is compared. In addition, PDs were loaded with the photoNORM and neodymium-sensitized upconverting nanoparticles (Nd-UCNPs). The Nd-UCNPs absorb NIR light at ∼800 nm and activate NO release from the trans-[Cr(PetA)(ONO)2]+ cation. The use of such ensembles as implants provides a potential strategy for the in vivo uncaging of NO at physiological targets triggered by tissue-transmitting NIR excitation. Also reported are the X-ray crystal structures of cis- and trans-{Cr(PetA)Cl2]Cl.
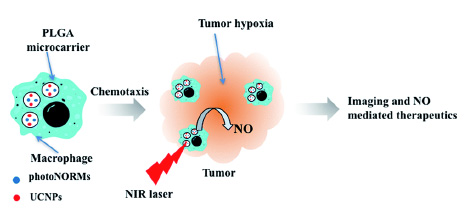
Macrophage-Mediated Delivery of Light Activated Nitric Oxide Prodrugs with Spatial, Temporal and Concentration Control
M. A. Evans, P.-J. Huang, Y. Iwamoto, K. N. Ibsen, E. M. Chan, Y. Hitomi, P. C. Ford, S. Mi-tragotri,
Chemical Science, 2018, 9, 3729-3741
DOI: 10.1039/c8sc00015h
Abstract
Nitric oxide (NO) holds great promise as a treatment for cancer hypoxia, if its concentration and lo-calization can be precisely controlled. Here, we report a “Trojan Horse” strategy to provide the nec-essary spatial, temporal, and dosage control of such drug-delivery therapies at targeted tissues. De-scribed is a unique package consisting of (1) a manganese–nitrosyl complex, which is a photoacti-vated NO-releasing moiety (photoNORM), plus Nd3+-doped upconverting nanoparticles (Nd-UCNPs) incorporated into (2) biodegradable polymer microparticles that are taken up by (3) bone-marrow derived murine macrophages. Both the photoNORM [Mn(NO)dpaqNO2]BPh4(dpaqNO2 = 2-[N,N-bis(pyridin-2-yl-methyl)]-amino-N′-5-nitro-quinolin-8-yl-acetamido) and the Nd-UCNPs are activat-ed by tissue-penetrating near-infrared (NIR) light at ∼800 nm. Thus, simultaneous therapeutic NO delivery and photoluminescence (PL) imaging can be achieved with a NIR diode laser source. The loaded microparticles are non-toxic to their macrophage hosts in the absence of light. The micropar-ticle-carrying macrophages deeply penetrate into NIH-3T3/4T1 tumor spheroid models, and when the infiltrated spheroids are irradiated with NIR light, NO is released in quantifiable amounts while emission from the Nd-UCNPs provides images of microparticle location. Furthermore, varying the intensity of the NIR excitation allows photochemical control over NO release. Low doses reduce lev-els of hypoxia inducible factor 1 alpha (HIF-1α) in the tumor cells, while high doses are cytotoxic. The use of macrophages to carry microparticles with a NIR photo-activated theranostic payload into a tumor overcomes challenges often faced with therapeutic administration of NO and offers the po-tential of multiple treatment strategies with a single system.
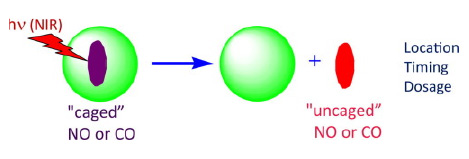
Metal complex strategies for photo-uncaging the small molecule bioregulators nitric oxide and carbon monoxide
P. C. Ford
Coord. Chem. Rev. 2018 376, 548-564.
DOI: 10.1016/j.ccr.2018.07.018
Abstract
Photochemical release (uncaging) of small molecule bioregulators (SMBs) such as nitric oxide (NO) or carbon monoxide (CO) at physiological sites offers exquisite control of timing, location and dos-age. However, photo-uncaging faces two major problems that challenge its therapeutic applications: the relatively poor transmision of visible light through tissue and the need to deliver the appropriate precursors to the desired targets. In this brief review are discussed research activities that address these issues of spatial-temporal control.
Dinuclear PhotoCORMs: Dioxygen-Assisted Carbon Monoxide Uncaging from Long-Wavelength-Absorbing Metal–Metal-Bonded Carbonyl Complexes
Li, Z.; Pierri, A. E.; Huang, P.-J.; Wu, G.; Iretskii, A. V.; Ford, P. C. Inorganic Chemistry 2017
DOI: 10.1021/acs.inorgchem.6b03138

Abstract
We describe a new strategy for triggering the photochemical release of caged carbon monoxide (CO) in aerobic media using long-wavelength visible and near-infrared (NIR) light. The dinuclear rhenium–manganese carbonyl complexes (CO)5ReMn(CO)3(L), where L = phenanthroline (1), bipyridine (2), biquinoline (3), or phenanthrolinecarboxaldehyde (4), each show a strong metal–metal-bond-to-ligand (σMM → πL*) charge-transfer absorption band at longer wavelengths. Photolysis with deep-red (1 and 2) or NIR (3 and 4) light leads to homolytic cleavage of the Re–Mn bonds to give mononuclear metal radicals. In the absence of trapping agents, these radicals primarily recombine to reform dinuclear complexes. In oxygenated media, however, the radicals react with dioxygen to form species much more labile toward CO release via secondary thermal and/or photochemical reactions. Conjugation of 4, with an amine-terminated poly(ethylene glycol) oligomer, gives a water-soluble derivative with similar photochemistry. In this context, we discuss the potential applications of these dinuclear complexes as visible/NIR-light-photoactivated CO-releasing moieties (photoCORMs).
A photoCORM nanocarrier for CO release using NIR light
Feb 2015
Pierri AE, Huang PJ, Garcia JV, Stanfill JG, Chui M, Wu G, Zheng N, & Ford PC. Chemical Communications 2015, 51, 2072-5.
DOI: 10.1039/C4CC06766E

Abstract
A water-soluble nanocarrier for a photo-activated CO releasing moiety (photoCORM) that can be triggered with NIR excitation is described. This has an upconversion nanoparticle core encapsulated by an amphiphilic polymer imparting both water solubility and a hydrophobic interior containing the photoCORM trans-Mn(bpy)(PPh3)2(CO)2. Such an ensemble offers a unique strategy for CO delivery to biol. targets.
Synthesis and Properties of Phosphine-Substituted Ruthenium(II) Polypyridine Complexes with Nitrogen Oxide
4. August 2015
Nakamura G, Kondo M, Crisalli M, Lee SK, Shibata A, Ford PC, & Masaoka S. Dalton Transactions, 2015, in press.
DOI: 10.1039/C5DT02994E
Abstract
Four novel phosphine-substituted ruthenium(II) polypyridine complexes with nitrogen oxides—trans(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (trans-NO2), cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (cis-NO2), [Ru(trpy)(dppbz)(NO2)]PF6 (PP-NO2), and cis(P,NO)-[Ru(trpy)(Pqn)(NO)](PF6)3 (cis-NO)—were synthesised (trpy = 2,2’:6’,2”-terpyridine, Pqn = 8-(diphenylphosphanyl)quinoline, and dppbz = 1,2-bis(diphenylphosphanyl)benzene). The influence of the number and position of the phosphine group(s) on the electronic structure of these complexes was investigated using single-crystal X-ray structural analysis, UV-vis absorption spectroscopy, and electrochemical measurements. The substitution lability of the nitrogen oxide ligand of each complex is discussed in comparison with that of the corresponding acetonitrile complexes.
Photoreactivity of a Quantum Dot-Ruthenium Nitrosyl Conjugate
18. Nov 2014
Franco LP, Cincillini SA, Biazzotto JC, Schiavon MA, Mikhailovsky A, Burks PT, Garcia JV, Ford PC, & Silva RS. Journal of Physical Chemistry A, 2014, 118, 12184-12191.
DOI: 10.1021/jp5111218
Abstract
 We describe the use of cadmium telluride quantum dots (CdTe QDs) as antennas for the photosensitization of nitric oxide release from a ruthenium nitrosyl complex with visible light excitation. The CdTe QDs were capped with mercaptopropionic acid to make them water-sol., and the ruthenium nitrosyl complex was cis-[Ru(NO)(4-ampy)(bpy)2]3+ (Ru-?NO; bpy is 2,2'-?bipyridine, and 4-ampy is 4-aminopyridine). Solns. of these two components demonstrated concn.-dependent quenching of the QD photoluminescence (PL) as well as photoinduced release of NO from Ru-NO when irradiated by 530 nm light. A NO release enhancement of ∼8 times resulting from this assocn. was obsd. under longer wavelength excitation in visible light range. The dynamics of the quenching detd. by both PL and transient absorption measurements were probed by ultrafast flash photolysis. A charge transfer mechanism is proposed to explain the quenching of the QD excited states as well as the photosensitized release of NO from Ru-NO.
We describe the use of cadmium telluride quantum dots (CdTe QDs) as antennas for the photosensitization of nitric oxide release from a ruthenium nitrosyl complex with visible light excitation. The CdTe QDs were capped with mercaptopropionic acid to make them water-sol., and the ruthenium nitrosyl complex was cis-[Ru(NO)(4-ampy)(bpy)2]3+ (Ru-?NO; bpy is 2,2'-?bipyridine, and 4-ampy is 4-aminopyridine). Solns. of these two components demonstrated concn.-dependent quenching of the QD photoluminescence (PL) as well as photoinduced release of NO from Ru-NO when irradiated by 530 nm light. A NO release enhancement of ∼8 times resulting from this assocn. was obsd. under longer wavelength excitation in visible light range. The dynamics of the quenching detd. by both PL and transient absorption measurements were probed by ultrafast flash photolysis. A charge transfer mechanism is proposed to explain the quenching of the QD excited states as well as the photosensitized release of NO from Ru-NO.
Photo-controlled release of NO and CO with inorganic and organometallic complexes.
Mar 2015
Pierri AE, Muizzi DA, Ostrowski AD, & Ford PC. Structure and Bonding, 2015, 165, 1-45.
DOI: 10.1007/430_2014_164
Abstract
The photochemical delivery of bioactive small molecules to physiological targets provides the opportunity to control the location, timing, and dosage of such delivery. We will discuss recent developments of the synthesis and studies of various metal complexes designed for targeted release of the bioregulatory diatomics nitric oxide and carbon monoxide. Of considerable interest are those systems where the NO or CO precursor and/or the photochemical product is luminescent such that imaging techniques allow one to identify the release location.
Photocatalytic Carbon Disulfide Production via Charge Transfer Quenching of Quantum Dots
12. Feb 2014 - JACS Cover
Bernt, CM; Burks, PT; DeMartino, AW; Pierri, AE; Levy, ES; Zigler, DF; Ford, PC. J. Am. Chem. Soc. 2014, 136(6), 2192-2195.
DOI: 10.1021/ja4083599
 Abstract
Abstract
Carbon disulfide, a potentially therapeutic small molecule, is generated via oxidative cleavage of 1,1-dithiooxalate (DTO) photosensitized by CdSe quantum dots (QDs). Irradiation of DTO–QD conjugates leads to ?irr independent photooxidation with a quantum yield of ~4% in aerated pH 9 buffersolution that drops sharply in deaerated solution. Excess DTO is similarly decomposed, indicating labile exchange at the QD surfaces and a photocatalyticcycle. Analogous photoreaction occurs with the O-tert-butyl ester tBuDTO in nonaqueous media. We propose that oxidation is initiated by hole transfer from photoexcited QD to surface DTO and that these substrates are a promising class of photocleavable ligands for modifying QD surface coordination
 as CS2 and CO2 are released.")
A video explaining the details of this intriguing process can be found in our photochemistry section, here.
This paper is also featured in Spotlights of Recent JACS Publications, J. Am. Chem. Soc, 2014, 136(6), 2177, found here.
UPDATE 2/18/14: This paper is featured as a podcast on JACS Beta, you can hear the interview with Tony DeMartino here, podcast # 48.
Nitric Oxide Releasing Materials Triggered by Near-Infrared Excitation through Tissue Filters
18. Nov 2013
Burks, PT; Garcia, JV; GonzalezIrias, R; Tillman, JT; Niu, M; Mikhailovsky, AA; Zhang, J; Zhang, F; Ford, PC. J. Am. Chem. Soc. 2013, Just accepted.
DOI: 10.1021/ja408516w
Abstract
 Novel materials for the photo-therapeutic release of the bioregulator nitric oxide (nitrogen monoxide) are described. Also reported is a method for scanning these materials with a focused NIR beam to induce photo-uncaging while minimizing damage from local heating. The new materials consist of poly(dimethylsiloxane) composites with near infrared-to-visible upconverting nanoparticles (UCNPs) that are cast into a biocompatible polymer disk (PD). These PDs are then impregnated with the photochemical nitric oxide precursor Roussin's black salt (RBS) to give UCNP_RBS_PD devices that generate NO when irradiated with 980 nm light. When the UCNP_RBS_PD composites were irradiated with NIR light through filters composed of porcine tissue, physiologically relevant NO concentrations were released, thus demonstrating the potential of such devices for minimally-invasive photo-therapeutic applications.
Novel materials for the photo-therapeutic release of the bioregulator nitric oxide (nitrogen monoxide) are described. Also reported is a method for scanning these materials with a focused NIR beam to induce photo-uncaging while minimizing damage from local heating. The new materials consist of poly(dimethylsiloxane) composites with near infrared-to-visible upconverting nanoparticles (UCNPs) that are cast into a biocompatible polymer disk (PD). These PDs are then impregnated with the photochemical nitric oxide precursor Roussin's black salt (RBS) to give UCNP_RBS_PD devices that generate NO when irradiated with 980 nm light. When the UCNP_RBS_PD composites were irradiated with NIR light through filters composed of porcine tissue, physiologically relevant NO concentrations were released, thus demonstrating the potential of such devices for minimally-invasive photo-therapeutic applications.
Multi-Photon Excitation in Uncaging the Small Molecule Bioregulator Nitric Oxide
17. Jun 2013
Garcia JV, Zhang F, & Ford PC. Philosophical Transactions of the Royal Society, A: Mathematical, Physical & Engineering Sciences, 2013, 371, 20120129 - .
Abstract
Multi-photon excitation allows one to use tissue transmitting near-infrared (NIR) light to access excited states with energies corresponding to single-photon excitation in the visible or ultraviolet wavelength ranges. Here, we present an overview of the application of both simultaneous and sequential multi-photon excitation in studies directed towards the photochemical delivery (‘uncaging’) of bioactive small molecules such as nitric oxide (NO) to physiological targets. Particular focus will be directed towards the use of dyes with high two-photon absorption cross sections and lanthanide ion-doped upconverting nanoparticles as sensitizers to facilitate the uncaging of NO using NIR excitation.
NIR-Triggered Release of Caged Nitric Oxide using Upconverting Nanostructured Materials
21. Dec 2012 - Small Cover

Garcia, JV; Yang, JP; Shen, DK; Yao, C; Li, XM; Wang, R; Stucky, GD; Zhao, DY; Ford PC; Zhang, F. Small. 2012, 8, 24, 3800-3805.
DOI: 10.1002/smll.201201213
Abstract
In this manuscript, a novel strategy for the therapeutic delivery of nitric oxide to physiological targets is described. Nitric oxide (NO) is a key vasodilator in mammalian cardiovascular systems and has been shown to sensitize tissue to gamma-radiation. However, photochemical NO precursors that can be activated by these near-infrared (NIR) wavelengths are extremely limited. Herein, we have addressed this issue by utilizing the known NIR to visible upconversion properties of lanthanide cations doped NaYF4 nanocrystals. This result is a potential game changer in multiphoton excitation based therapeutic delivery of NO and other small molecule bioregulators.
Bioinorganic Chemistry of Small Molecule Bioregulators (SMBs) Papers
Dinitrosyl Iron Complexes (DNICs). From Spontaneous Assembly to Biological Roles
D. R. Truzzi, N. M. Medeiros, O. Augusto, P. C. Ford
Inorganic Chemistry 2021
DOI: 10.1021/acs.inorgchem.1c00823
Abstract
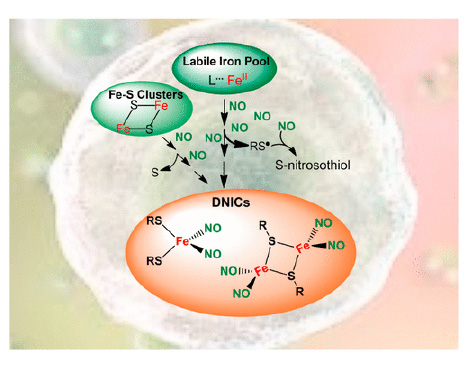
Dinitrosyl iron complexes (DNICs) are spontaneously and rapidly generated in cells. Their assem-bly requires nitric oxide (NO), biothiols, and nonheme iron, either labile iron or iron–sulfur clus-ters. Despite ubiquitous detection by electron paramagnetic resonance in NO-producing cells, the DNIC’s chemical biology remains only partially understood. In this Forum Article, we address the reaction mechanisms for endogenous DNIC formation, with a focus on a labile iron pool as the iron source. The capability of DNICs to promote S-nitrosation is discussed in terms of S-nitrosothiol generation associated with the formation and chemical reactivity of DNICs. We also highlight how elucidation of the chemical reactivity and the dynamics of DNICs combined with the development of detection/quantification methods can provide further information regarding their participation in physiological and pathological processes.
Nitric Oxide Dioxygenation by O2 Adducts of Manganese Porphyrins
T. S. Kurtikyan, V.A. Hayrapetyan, A. A. Hovhannisyan, G. G. Martirosyan, G. Sh. Hovhannisyan, A. V. Iretski, P. C. Ford
Inorganic Chem, 2020, 59, 17224–17233
DOI: 10.1021/acs.inorgchem.0c02464
Abstract
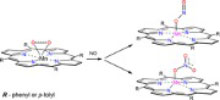
We describe here nitric oxide dioxygenation (NOD) by the dioxygen manganese porphyrin adducts Mn(Por)(η2-O2) (Por2– = the meso-tetra-phenyl or meso-tetra-p-tolylporphyrinato dianions, TPP2– and TTP2–). The Mn(Por)(η2-O2) was assembled by adding O2 to sublimed layers of MnII(Por). When NO was introduced and the temperature was slowly raised from 80 to 120 K, new IR bands with correlated intensities grew concomitant with depletion of the υ(O2) band. Isotope labeling ex-periments with 18O2, 15NO, and N18O combined with DFT calculations provide the basis for identi-fying the initial intermediates as the six-coordinate peroxynitrito complexes (ON)Mn(Por)(η1-OONO). Further warming to room temperature led to formation of the nitrato complexes Mn(Por)(η1-ONO2), thereby demonstrating the ability of these metal centers to promote NOD. How-ever, comparable quantities of the nitrito complexes Mn(Por)(η1-ONO) are also formed. In contrast, when the analogous reactions were initiated with the weak σ-donor ligand tetrahydrofuran or dime-thyl sulfide present in the layers, formation of Mn(Por)(η1-ONO2) is strongly favored (∼90%). The latter are formed via a 6-coordinate intermediate (L)Mn(Por)(η1-ONO2) (L = THF or DMS) that los-es L upon warming. These reaction patterns are compared to those observed previously with analo-gous iron and cobalt porphyrin complexes.

The Solution Chemistry of Nitric Oxide and Other Reactive Nitrogen Species
P. C. Ford, K. M. Miranda,
Nitric Oxide, 2020, 103, 31-46.
DOI: 10.1016/j.niox.2020.07.004
Abstract
In this article we discuss the fundamental chemical and physical properties of NO and related nitrogen oxides (NO2−, NO2, N2O3, etc.) under solution conditions relevant to mammalian biology.
Thiyl Radicals Are Co-products of Dinitrosyl Iron Complexes (DNICs) Formation
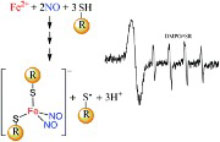
D. R. Truzzi, O. Augusto, P. C. Ford
Chemical Communications 2019, 55, 9156-9159.
DOI: 10.1039/C9CC04454J
Abstract
Thiyl radicals are detected by EPR as co-products of dinitrosyl iron complex (DNIC) formation. In demonstrating that DNIC formation generates RS˙ in a NO rich environment, these results provide a novel route for S-nitroso thiol formation.
Dynamics of Dinitrosyl Iron Complex (DNIC) Formation with Low Molecular Weight Thiols
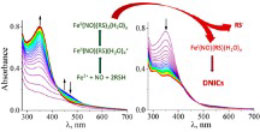
D. R. Truzzi, O. Augusto, A. V. Iretskii, P. C. Ford,
Inorganic Chemistry, 2019, 58, 13446-13456.
DOI: 10.1021/acs.inorgchem.9b02338
Abstract
Dinitrosyl iron complexes (DNICs) are ubiquitous in mammalian cells and tissues producing nitric oxide (NO) and have been argued to play key physiological and pathological roles. Nonetheless, the mechanism and dynamics of DNIC formation in aqueous media remain only partially understood. Here, we report a stopped-flow kinetics and density functional theory (DFT) investigation of the reaction of NO with ferrous ions and the low molecular weight thiols glutathione (GSH) and cysteine (CysSH) as well as the peptides WCGPC and WCGPY to produce DNICs in pH 7.4 aqueous media. With each thiol, a two-stage reaction pattern is observed. The first stage involves several rapidly established pre-equilibria leading to a ferrous intermediate concluded to have the composition FeII(NO)(RS)2(H2O)x(C). In the second stage, C undergoes rate-limiting, unimolecular autoreduction to give thiyl radical (RS•) plus the mononitrosyl Fe(I) complex FeI(NO)(RS)(H2O)xfollowing the reactivity order of CysSH > WCGPC > WCGPY > GSH. Time course simulations using the experimentally determined kinetics parameters demonstrate that, at a NO flux characteristic of inflammation, DNICs will be rapidly formed from intracellular levels of ferrous iron and thiols. Furthermore, the proposed mechanism offers a novel pathway for S-nitroso thiol (RSNO) formation in a biological environment.
Carbon disulfide. Just toxic or also bioregulatory and/or therapeutic?
A. W. DeMartino, D. F. Zigler, J. M. Fukuto, P C. Ford
Chemical Society Reviews 2017, 46, 21-39.
DOI: 10.1039/C6CS00585C
Abstract

The overview presented here has the goal of examining whether carbon disulfide (CS2) may play a role as an endogenously generated bioregulator and/or has therapeutic value. The neuro- and repro-ductive system toxicity of CS2 has been documented from its long-term use in the viscose rayon industry. CS2 is also used in the production of dithiocarbamates (DTCs), which are potent fungi-cides and pesticides, thus raising concern that CS2 may be an environmental toxin. However, DTCs also have recognized medicinal use in the treatment of heavy metal poisonings as well as having po-tency for reducing inflammation. Three known small molecule bioregulators (SMBs) nitric oxide, carbon monoxide, and hydrogen sulfide were initially viewed as environmental toxins. Yet each is now recognized as having intricate, though not fully elucidated, biological functions at concentration regimes far lower than the toxic doses. The literature also implies that the mammalian chemical bi-ology of CS2 has broader implications from inflammatory states to the gut microbiome. On these bases, we suggest that the very nature of CS2 poisoning may be related to interrupting or over-whelming relevant regulatory or signaling process(es), much like other SMBs.
Uncaging Carbon Disulfide. Delivery Platforms for Potential Pharmacological Applications: A Mechanistic Approach
A. W. DiMartino, M. L. Souza, P. C. Ford
Chemical Science, 2017, 8, 7186 – 7196.
DOI: 10.1039/c7sc02727c
Abstract
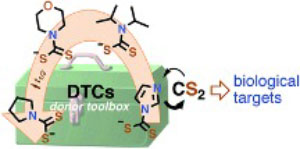
We describe the kinetics of the formation and decay of a series of dithiocarbamates under physio-logical conditions. The goal is to provide a toolbox of compounds that release CS2 by well-defined kinetics in such media. Carbon disulfide is a known environmental toxin, but there is fragmentary evidence suggesting that CS2 may have bioregulatory and/or therapeutic roles in mammalian biology. Further investigation of such roles will require methodologies for controlled delivery of this bioac-tive small molecule to specific targets. Reported here are mechanistic and computational studies of CS2 release from a series of dithiocarbamate anions (DTCs), where R2N represents several different secondary amido groups. The various DTCs under physiologically relevant conditions show a tre-mendous range of reactivities toward CS2 dissociation with decay lifetimes ranging from ∼2 s for imidazolidyldithiocarbamate (ImDTC−) to ∼300 s for diisopropyldithiocarbamate (DIDTC−) to >24 h for pyrrolidinyldithiocarbamate (PDTC−) in pH 7.4 phosphate buffer solution at 37 °C. Thus, by making the correct choice of these tools, one can adjust the flux of CS2 in a biological experiment, while the least reactive DTCs could serve as controls for evaluating the potential effects of the dithi-ocarbamate functionality itself. Kinetics studies and density functional calculations are used to probe the mechanism of DTC− decay. In each case, the rate of CS2 dissociation is acid dependent; however, the DFT studies point to a mechanistic pathway for ImDTC− that is different than those for DIDTC−. The role of general acid catalysis is also briefly probed.
Biological thiols and carbon disulfide. The formation and decay of trithiocarbonates under physiologically relevant conditions
M. Lima Souza, A.W. DeMartino, P. C. Ford
ACS Omega, 2017, 2, 6535-6543,
Abstract
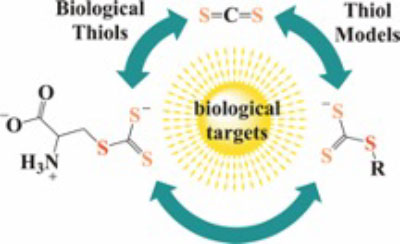
Carbon disulfide is an environmental toxin, but there are suggestions in the literature that it may also have regulatory and/or therapeutic roles in mammalian physiology. Thiols or thiolates would be likely biological targets for an electrophile, such as CS2, and in this context, the present study exam-ines the dynamics of CS2 reactions with various thiols (RSH) in physiologically relevant near-neutral aqueous media to form the respective trithiocarbonate anions (TTC–, also known as “thi-oxanthate anions”). The rates of TTC– formation are markedly pH-dependent, indicating that the reactive form of RSH is the conjugate base RS–. The rates of the reverse reaction, that is, decay of TTC– anions to release CS2, is pH-independent, with rates roughly antiparallel to the basicities of the RS–conjugate base. These observations indicate that the rate-limiting step of decay is simple CS2 dissociation from RS–, and according to microscopic reversibility, the transition state of TTC– formation would be simple addition of the RS–nucleophile to the CS2 electrophile. At pH 7.4 and 37 °C, cysteine and glutathione react with CS2 at a similar rate but the trithiocarbonate product under-goes a slow cyclization to give 2-thiothiazolidine-4-carboxylic acid. The potential biological rele-vance of these observations is briefly discussed
Dinitrosyl Iron Complexes with Cysteine. Kinetics Studies of the Formation and Reactions of DNICs in Aqueous Solution
5. Dec 2014
Pereira JCM, Iretskii AV, Han RM, & Ford, PC. J. Am. Chem. Soc. 2015, 137, 328-336.
DOI: 10.1021/ja510393q
Abstract

Kinetics studies provide mechanistic insight regarding the formation of dinitrosyl iron complexes (DNICs), which are now viewed as playing important roles in the mammalian chem. biol. of the ubiquitous bioregulator nitric oxide (NO). Reactions in deaerated aq. solns. containing FeSO4, cysteine (CysSH), and NO demonstrate that both the rates and the outcomes are markedly pH dependent. The dinuclear DNIC Fe2(u-CysS)2(NO)4, a Roussin's red salt ester (Cys-RSE), is formed at pH 5.0 as well as at lower concns. of cysteine in neutral pH solns. The mononuclear DNIC Fe(NO)2(CysS)2- (Cys-DNIC) is produced from the same three components at pH 10.0 and at higher cysteine concentrations at neutral pH. The kinetics studies suggest that both Cys-RSE and Cys-DNIC are formed via a common intermediate Fe(NO)(CysS)2-. Cys-DNIC and Cys-RSE interconvert, and the rates of this process depend on the cysteine concn. and on the pH. Flash photolysis of the Cys-RSE formed from Fe(II)/NO/cysteine mixts. in anaerobic pH 5.0 soln. led to reversible NO dissocn. and a rapid, second-?order back reaction with a rate const. kNO = 6.9 × 107 M-1 s-1. In contrast, photolysis of the mononuclear-DNIC species Cys-DNIC formed from Fe(II)/NO/cysteine mixts. in anaerobic pH 10.0 soln. did not labilize NO but instead apparently led to release of the CysS• radical. These studies illustrate the complicated reaction dynamics interconnecting the DNIC species and offer a mechanistic model for the key steps leading to these non-heme iron nitrosyl complexes.
Six-coordinate Nitrito and Nitrato Complexes of Manganese Porphyrin
4. Nov 2014
Kurtikyan TS, Hayrapetyan VA, Mehrabyan MM, & Ford PC. Inorganic Chemistry, 2014, 53, 11948-11959.
DOI: 10.1021/ic5014329
Abstract
Reaction of small increments of NO2 gas with sublimed amorphous layers of MnII(TPP) (TPP = meso-tetra-phenylporphyrinato dianion) in a vacuum cryostat leads to formation of the 5-coordinate monodentate nitrato complex MnIII(TPP)(1-ONO2) (II). This transformation proceeds through the two distinct steps with initial formation of the five coordinate O-nitrito complex MnIII(TPP)(1-ONO) (I) as demonstrated by the electronic absorption spectra and by FTIR spectra using differently labeled nitrogen dioxide. A plausible mechanism for the second stage of reaction is offered based on the spectral changes observed upon subsequent interaction of 15NO2 and NO2 with the layered Mn(TPP). Low-temperature interaction of I and IIwith the vapors of various ligands L (L = O-, S-, and N-donors) leads to formation of the 6-coordinate O-nitrito MnIII(TPP)(L)(1-ONO) and monodentate nitrato MnIII(TPP)(L)(1-ONO2) complexes, respectively. Formation of the 6-coordinate O-nitrito complex is accompanied by the shifts of the (NO) band to lower frequency and of the (N–O) band to higher frequency. The frequency difference between these bands ?? = (NO) – (N–O) is a function of L and is smaller for the stronger bases. Reaction of excess NH3 with I leads to formation of Mn(TPP)(NH3)(?1-ONO) and of the cation [Mn(TPP)(NH3)2]+ plus ionic nitrite. The nitrito complexes are relatively unstable, but several of the nitrato species can be observed in the solid state at room temperature. For example, the tetrahydrofuran complex Mn(TPP)(THF)(1-ONO2) is stable in the presence of THF vapors (∼5 mm), but it loses this ligand upon high vacuum pumping at RT. When L = dimethylsulfide (DMS), the nitrato complex is stable only to ~−30 °C. Reactions of II with the N-donor ligands NH3, pyridine, or 1-methylimidazole are more complex. With these ligands, the nitrato complexes MnIII(TPP)(L)(1-ONO2) and the cationic complexes [Mn(TPP)(L)2]+ coexist in the layer at room temperature, the latter formed as a result of NO3–displacement when L is in excess.
Reaction of a Bridge Frustrated Lewis Pair with Nitric Oxide: A Kinetics Study
5. Jan 2014
Pereira, JCM; Sajid, M; Kehr, G; Wright, AM; Schirmer, B; Qu, ZW; Grimme, S; Erker, G; Ford, PC. J. Am. Chem Soc. 2014, 136(1), 513-519.
DOI: 10.1021/ja4118335
Abstract
 reaction with nitric oxide") Described is a kinetics and computational study of the reaction of NO with the intramolecular bridged P/B frustrated Lewis pair (FLP) endo-2-(dimesitylphosphino)-exo-3-bis(pentafluorophenyl)boryl-norbornane to give a persistent FLP-NO aminoxyl radical. This reaction follows a second-order rate law, first-order in [FLP] and first-order in [NO], and is markedly faster in toluene than in dichloromethane. By contrast, the NO oxidation of the phosphine base 2-(dimesitylphosphino)norbornene to the corresponding phosphine oxide follows a third-order rate law, first-order in [phosphine] and second-order in [NO]. Formation of the FLP-NO radical in toluene occurs with a ?H
Described is a kinetics and computational study of the reaction of NO with the intramolecular bridged P/B frustrated Lewis pair (FLP) endo-2-(dimesitylphosphino)-exo-3-bis(pentafluorophenyl)boryl-norbornane to give a persistent FLP-NO aminoxyl radical. This reaction follows a second-order rate law, first-order in [FLP] and first-order in [NO], and is markedly faster in toluene than in dichloromethane. By contrast, the NO oxidation of the phosphine base 2-(dimesitylphosphino)norbornene to the corresponding phosphine oxide follows a third-order rate law, first-order in [phosphine] and second-order in [NO]. Formation of the FLP-NO radical in toluene occurs with a ?H of 13 kcal mol–1, a feature that conflicts with the computation-based conclusion that NO addition to a properly oriented B/P pair should be nearly barrierless. Since the calculations show the B/P pair in the most stable solution structure of this FLP to have an unfavorable orientation for concerted reaction, the observed barrier is rationalized in terms of the reversible formation of a [B]-NO complex intermediate followed by a slower isomerization–ring closure step to the cyclic aminoxyl radical. This combined kinetics/theoretical study for the first time provides insight into mechanistic details for the activation of a diatomic molecule by a prototypical FLP.
Nitrite reduction mediated by heme models. Routes to NO and HNO?
19. Feb 2013
Heinecke JL, Khin C, Pereira JCM, Suárez SA, Iretskii AV, Doctorovich F, & Ford PC. J. Am. Chem. Soc. 2013, 135, 4007-4017.
DOI: 10.1021/ja312092x
Abstract
The water-soluble ferriheme model FeIII(TPPS) mediates oxygen atom transfer from inorganic nitrite to a water soluble phosphine (tppts), dimethyl sulfide, and the biological thiols cysteine (CysSH) and glutathione (GSH). The products with the latter reductant are the respective sulfenic acids CysS(O)H and GS(O)H, although these reactive intermediates are rapidly trapped by reaction with excess thiol. The nitrosyl complex FeII(TPPS)(NO) is the dominant iron species while excess substrate is present. However, in slightly acidic media (pH ~6), the system does not terminate at this very stable ferrous nitrosyl. Instead it displays a matrix of redox transformations linking spontaneous regeneration of FeIII(TPPS) to the formation of both N2O and NO. Electrochemical sensor and trapping experiments demonstrate that HNO (nitroxyl) is formed, at least when tppts is the reductant. HNO is the likely predecessor of the N2O. A key pathway to NO formation is nitrite reduction by FeII(TPPS), and the kinetics of this iron-mediated transformation are described. Given that inorganic nitrite has protective roles during ischemia/reperfusion (I/R) injury to organs, attributed in part to NO formation, and that HNO may also reduce net damage from I/R, the present studies are relevant to potential mechanisms of such nitrite protection.
New Zn(II) complexes with N2S2 Schiff Base Ligands. Experimental and Theoretical Studies of the Role of Zn(II) in Disulfide-Thiolate Exchange.
29. Mar 2014
Amirnasr M, Bagheri M, Farrokhpour H, Schenk KJ, Mereiter K, & Ford PC. 2014, 136, 513-519.
DOI: 10.1021/ja4118335
 complexes with N2S2 Schiff base ligands") Abstract
Abstract
Described are the synthesis and characterization of two, potentially tetradentate, N2S2 Schiff-base ligands, containing a disulfide bond, N,N′-bis(3-phenylprop-2-en-1-ylidene)-2,2′-disulfanediyldianiline (L1) and N,N′-bis(3,3-diphenylprop-2-en-1-ylidene)-2,2′-disulfanediyldianiline (L2), and their reaction with Zn2+. Surprisingly, both L1 and L2 undergo reductive disulfide bond scission upon reaction with Zn2+ in alcoholic media to give, under alcohol oxidation, the respective Zn(NS)2 complexes Zn(L3)2 (1) and Zn(L4)2 (2), where the L3 and L4 are the respective bidentate thiolate-imine anions. The ligands L1 and L2 and the complexes 1 and 2 have been characterized spectroscopically, and the crystal and molecular structures of the two complexes have been determined by single crystal X-ray diffraction. The coordination geometry around Zn(II) centers in both complexes is a distorted tetrahedron. In addition, DFT calculations (B3LYP/LANL2DZ/6-311++G(d,p)) support the structure of 1. Cyclic voltammetric studies demonstrate that Zn(II) shifts the reduction potential of the disulfide ligands L1 and L2 to less negative values thus making them more susceptible to reductive cleavage of the disulfide bond. The results of semi-empirical PM6 calculations offer key insight into the nature of the transition state for this reaction.
Tracking Reactive Intermediates by FTIR Monitoring of Reactions in Low-Temperature Sublimed Solids. Nitric Oxide Disproportionation Mediated by the Ruthenium(II) Carbonyl Porphyrin Ru(TPP)(CO)
13. Apr 2013
Azizyan AS, Kurtikyan TS, Martirosyan GG, & Ford PC. Inorg. Chem. 2013, 52, 5201-5205.
DOI: 10.1021/ic400102q
 Abstract
Abstract
Interaction of NO (15NO) with amorphous layers of Ru(II) carbonyl porphyrin was monitored by FTIR spectroscopy from 80 K to room temperature. An intermediate spectrally characterized at very low temperatures (110 K) with ?(CO) at 2001 cm−1 and (NO) at 1810 (1777 cm−1 for 15NO isotopomer) was readily assigned to the mixed carbonyl−nitrosyl complex Ru(TPP)(CO)(NO), which is the logical precursor to CO labilization.
Weak coordination of neutral S- and O-donor proximal ligands to a ferrous porphyrin nitrosyl. Characterization of 6-Coordinate Complexes at Low T.
Apr 2013
Martirosyan GG, Kurtikyan TS, Hovhannisyan AA, Iretskii A, & Ford PC. J. Inorg. Biochem. 2013, 121, 129-133.
DOI: 10.1016/j.jinorgbio.2012.12.017
Abstract
The interaction of the S- and O-donor ligands tetrahydrothiophen (THT) and tetrahydrofuran (THF) with the ferrous nitrosyl complex Fe(TTP)(NO) (TTP2 − is meso-tetra-p-tolyl-porphyrinatodianion) was studied at various temperatures both in solid state and solution using electronic and infrared absorption spectroscopy. Upon addition of these ligands to a cryostat containing sublimed layers of Fe(TTP)(NO), no complex formation was detected at room temperature. However, upon lowering the temperature, spectral changes were observed that are consistent with ligand binding in axial position trans to the NO (the proximal site) and formation of the six-coordinate adducts. Analogous behavior was observed in solution. In both media, the six-coordinate adducts are stable only at low temperature and dissociate to the 5-coordinate nitrosyl complexes upon warming.

The NO stretching frequencies of the six-coordinate thioether and ether complexes were recorded and binding constants for the weak bonding of proximal THF and THT ligands were determined from the spectral changes. These parameters are compared with those obtained for the N-donor ligand pyrrolidine.
Mechanisms of Nitric Oxide Reactions Mediated by Biologically Relevant Metal Centers
5. Oct 2013
Ford PC, Pereira JCM, & Miranda KM. Structure and Bonding, 2013, book chapter.
DOI: 10.1007/430_2013_117
Abstract
Here, we present an overview of mechanisms relevant to the formation and several key reactions of nitric oxide (nitrogen monoxide) complexes with biologically relevant metal centers. The focus will be largely on iron and copper complexes. We will discuss the applications of both thermal and photochemical methodologies for investigating such reactions quantitatively.
Sustainable Chemistry and Biomass Conversion Papers
Catalysis in Biomass Conversion

Advances in Inorganic Chemistry, Volume 77
P.C. Ford, R.van Edik, co-Editors.
Elsevier, 2021
ISBN: 978-0-323-85058-2
Summary: The 19th Century was the century of coal while the 20th Century was the century of oil and natural gas. It is inevitable that the 21st Century will be the century of renewable energy and feedstocks. For example, Lignocellulose, the principal component of woody biomass, is a sustainable feedstock that has the potential to be the largest renewable resource on which we can rely for producing the commodity chemicals and liquid transportation fuels needed for a carbon-neutral society. It is non-edible and is self-produced in large scale world-wide by the plant growth in the biosphere. Catalytic biomass conversion should play a significant role in this societal transformation. This is a very broad topic given the marked diversity of biomass-based resources, but real progress is being made in addressing the key challenges. This ten chapter volume brought together highly respected experts on biomass conversion to discuss their approaches to upgrading biomass components to the renewable fuels and chemicals for societal needs in the future.
One-pot Hydrodeoxygenation (HDO) of Lignin Monomers to C9 Hydrocarbons co-catalysed by Ru/C and Nb2O5
S. Li, B. Liu, J. Truong, Z. Luo, P. C. Ford, M. Abu-Omar
Green Chemistry. 2020, 22, 7406-7416
DOI: 10.1039/d0gc01692f
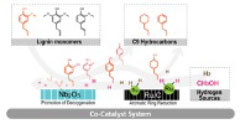
A physical mixture of Ru/C and Nb2O5 is an effective catalyst for upgrading lignin monomers under low H2 pressure at 250 °C to a clean cut of hydrocarbon liquid fuels. The reaction solvent is water with a small amount of methanol additive. Hydrodeoxygenation (HDO) was evaluated using dihy-droeugenol (DHE) as an exemplary lignin monomer model. Under optimized conditions, 100% conversion of DHE and very high selectivity to propyl cyclohexane (C9 hydrocarbon) was achieved. Nb2O5 was prepared at a low temperature (450 °C) and was shown to contain acid sites that enhance the production of fully deoxygenated products. The methanol additive serves as a hy-drogen source for the Ru/C catalysed reduction of the aromatic ring. In addition, when a substrate mixture of DHE, isoeugenol and 4-allylsyringol simulating lignin products was employed, 100% conversion to propyl cyclohexane (76%) and propyl benzene (24%) was observed, thereby suggest-ing the general applicability of this catalyst system for funneling lignin monomers into a clean cut of hydrocarbon liquid fuels. This study sheds light on the function of each catalyst component and provides a simple and green utilization of biomass monomers as a feedstock for renewable hydro-carbon fuels.
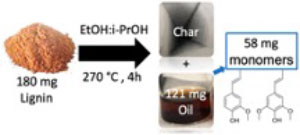
Hydrogenolysis of Organosolv Lignin in Ethanol/Isopropanol Media without Added Transi-tion-Metal Catalyst
C. Cheng, J. Truong, J. A. Barrett, D. Shen, M. M. Abu-Omar, P. C. Ford
ACS Sustainable Chem. Eng. 2020, 8, 1023−1030.
DOI: 10.1021/acssuschemeng.9b05820
Abstract
Lignin is the largest renewable source of aromatic chemical building blocks on the planet and has great potential for the production of value-added chemicals. Herein, we describe lignin hydrogenol-ysis/depolymerization of organosolv poplar lignin (OPL) in ethanol/isopropanol solvent in the ab-sence of added catalysts. Different f/i-PrOH ratios as well as various reaction conditions were evaluated. OPL depolymerization was more effective in the mixed media than in ethanol or isopro-panol alone. Heating OPL at 270 °C for 4 h in 50:50 (v:v) EtOH/i-PrOH in a closed pressure vessel gave an overall oil yield of 70 wt %, of which about 48% consisted of the monomers (E)-4-propenyl syringol and isoeugenol. Notably, these catalyst-free reactions in ethanol/isopropanol me-dia show monomer yields comparable to those reported for lignin depolymerization using precious metal catalysts and dihydrogen, which suggests unexpectedly favorable H-donor ability of this mixed alcohol medium.
Pinch of Salt Improves n-Butanol Selectivity in the Guerbet Condensation of Ethanol over Cu-doped Mg/Al Oxides
J. A. Barrett, Z. R. Jones, Cr. Stickelmaier, N. Schopp, P. C. Ford
ACS Sustainable Chem. Eng., 2018, 6, 15119-15126.
https://pubs.acs.org/doi/10.1021/acssuschemeng.8b03589
Abstract
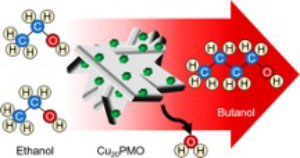
Improvement of processes that utilize renewable feedstocks to produce chemicals ordinarily derived from fossil carbon feedstocks is paramount to creating environmentally sustainable chemical and fuel industries. Catalytic conversion of biorefinery-derived ethanol via the Guerbet condensation has the potential to serve such a purpose. In this contribution, we demonstrate marked selectivity changes in the ethanol condensation over a copper-doped porous metal oxide catalyst upon a single exposure to ppm quantities of a soluble chloride source. Without this modification, ethyl acetate is the major product, with the others being n-butanol (principally) plus some 1-hexanol, butyl acetate, and ethyl butanoate. A single exposure to chloride increases activity and, more importantly, dramatically changes selectivity to give n-butanol and 1-hexanol as the largest products. X-ray diffraction and basicity measurements pre- and post-reaction were used to examine how chloride alters the properties of this Earth-abundant catalyst prepared by calcining a Cu-doped Mg/Al hydrotalcite.
Temperature Tuning the Catalytic Reactivity of Cu-Doped Porous Metal Oxides with Lignin Models
C. M. Bernt, H. Manesewan, M. Chui, M. Boscolo, P. C. Ford
ACS Sustainable Chem. Eng., 2018, 6, 2510–2516.
DOI: https://pubs.acs.org/doi/abs/10.1021/acssuschemeng.7b03969
Abstract
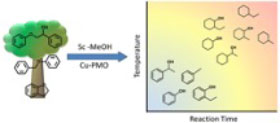
Reported are the temperature dependencies of the temporal product evolution for lignin model com-pounds over copper-doped porous metal oxide (CuPMO) in supercritical-methanol (sc-MeOH). These studies investigated 1-phenylethanol (PPE), benzyl phenyl ether (BPE), dihydrobenzofuran (DHBF), and phenol over operating temperature ranges from 280 to 330 °C. The first three model compounds represent the β-O-4 and α-O-4 linkages in lignin as well as the furan group commonly found in the β-5 linkage. Phenol was investigated due to its key role in product proliferation as not-ed in earlier studies with this Earth-abundant catalyst. In general, the apparent activation energies for ether hydrogenolysis proved to be significantly lower than that for phenol hydrogenation, a major side reaction leading to product proliferation. Thus, temperature tuning is a promising strategy to preserve product aromaticity as demonstrated by the more selective conversion of BPE and PPE at lower temperatures. Rates of methanol reforming over CuPMO were also studied over the tempera-ture range of 280–320 °C since it is this process that generates the reducing equivalents for this cata-lytic system. In the absence of substrate, the gaseous products H2, CO, and CO2 were formed in ra-tios stoichiometrically consistent with catalyzed methanol reformation and water gas shift reactions. The latter studies suggest that the H2 production ceases to be rate limiting early in batch reactor ex-periments but also suggest that H2overproduction may contribute to product proliferation.
Probing the Lignin Disassembly Pathways with Modified Catalysts Based on Cu-Doped Porous Metal Oxides
7 March 2017
Chui, M.; Metzker, G.; Bernt, C.M.; Tran, A.;Burtoloso A.C.; Ford, P.C.
DOI: 10.1021/acssuschemeng.6b02954
Abstract
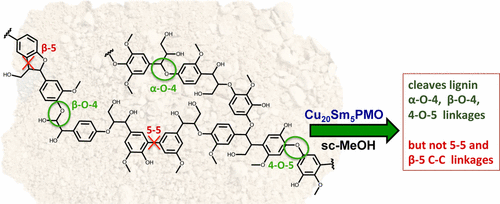
Described are the selectivities observed for reactions of lignin model compounds with modifications of the copper-doped porous metal oxide (CuPMO) system previously shown to be a catalyst for lignin disassembly in supercritical methanol (Matson et al., J. Amer. Chem. Soc. 2011, 133, 14090–14097). The models studied are benzyl phenyl ether, 2-phenylethyl phenyl ether, diphenyl ether, biphenyl, and 2,3-dihydrobenzofuran, which are respective mimetics of the ?-O-4, ?-O-4, 4-O-5, 5-5, and ?-5 linkages characteristic of lignin. Also, briefly investigated as a substrate is poplar organosolv lignin. The catalyst modifications included added samarium(III) (both homogeneous and heterogeneous) or formic acid. The highest activity for the hydrogenolysis of aryl ether linkages was noted for catalysts with Sm(III) incorporated into the solid matrix of the PMO structure. In contrast, simply adding Sm3+ salts to the solution suppressed the hydrogenolysis activity. Added formic acid suppressed aryl ether hydrogenolysis, presumably by neutralizing base sites on the PMO surface but at the same time improved the selectivity toward aromatic products. Acetic acid induced similar reactivity changes. While these materials were variously successful in catalyzing the hydrogenolysis of the different ethers, there was very little activity toward the cleavage of the 5-5 and ?-5 C-C bonds that represent a small, but significant, percentage of the linkages between monolignol units in lignins.
Enhancing Aromatic Production from Reductive Lignin Disassembly: in situ O-methylation of Phenolic Intermediates.
 15 Sept 2016
15 Sept 2016
Barrett, J. A.; Gao, Y.; Bernt, C. M.; Chui, M.; Tran, A. T.; Foston, M. B.; Ford, P. C.
DOI: 10.1021/acssuschemeng.6b01827
Abstract
The selective conversion of lignin into aromatic compounds has the potential to serve as a “green” alternative to the production of petrochemical aromatics. Herein, we evaluate the addition of dimethyl carbonate (DMC) to a biomass conversion system that uses a Cu-doped porous metal oxide (Cu20PMO) catalyst in supercritical methanol (sc-MeOH) to disassemble lignin with little to no char formation. While Cu20PMO catalyzes C–O hydrogenolysis of aryl–ether bonds linking lignin monomers, it also catalyzes arene methylation and hydrogenation, leading to product proliferation. The MeOH/DMC co-solvent system significantly suppresses arene hydrogenation of the phenolic intermediates responsible for much of the undesirable product diversity via O-methylation of phenolic −OH groups to form more stable aryl-OCH3 species. Consequently, product proliferation was greatly reduced and aromatic yields greatly enhanced with lignin models, 2-methoxy-4-propylphenol, benzyl phenyl ether, and 2-phenoxy-1-phenylethan-1-ol. In addition, organosolv poplar lignin (OPL) was examined as a substrate in the MeOH/DMC co-solvent system. The products were characterized by nuclear magnetic resonance spectroscopy (31P, 13C, and 2D 1H–13C NMR) and gas chromatography–mass spectrometry techniques. The co-solvent system demonstrated enhanced yields of aromatic products.
Mapping reactivities of aromatic models with a lignin disassembly catalyst. Steps toward controlling product selecivity.
15 Nov 2015
Bernt, C. M., Bottari, G., Barrett, J. A., Scott, S. L., Barta, K., Ford P.C.
DOI: 10.1039/C5CY01555C
Abstract

Catalytic Conversion of Non-food Woody Biomass Solids to Organic Liquids
18. Apr 2014
Barta K & Ford PC. Acc. Chem. Res. 2014, 47, 1503-1512.
DOI: 10.1021/ar4002894
Abstract
 This Account outlines recent efforts in our laboratories addressing a fundamental challenge of sustainability chemistry, the effective utilization of biomass for production of chemicals and fuels. Efficient methods for converting renewable biomass solids to chemicals and liquid fuels would reduce society's dependence on non-renewable petroleum resources while easing the atmospheric carbon dioxide burden. However, the major non-food component of biomass is lignocellulose, a matrix of the biopolymers cellulose, hemicellulose and lignin. New approaches are needed to effect facile conversion of lignocellulose solids to liquid fuels and to other chemical precursors without the formation of intractable side products and with sufficient specificity to give economically sustainable product streams.
This Account outlines recent efforts in our laboratories addressing a fundamental challenge of sustainability chemistry, the effective utilization of biomass for production of chemicals and fuels. Efficient methods for converting renewable biomass solids to chemicals and liquid fuels would reduce society's dependence on non-renewable petroleum resources while easing the atmospheric carbon dioxide burden. However, the major non-food component of biomass is lignocellulose, a matrix of the biopolymers cellulose, hemicellulose and lignin. New approaches are needed to effect facile conversion of lignocellulose solids to liquid fuels and to other chemical precursors without the formation of intractable side products and with sufficient specificity to give economically sustainable product streams.
We have devised a novel catalytic system whereby the renewable feedstocks cellulose, organosolv lignin and even lignocellulose composites such as sawdust are transformed into organic liquids. The reaction medium is supercritical methanol (sc-MeOH), while the catalyst is a copper-doped porous metal oxide (PMO) prepared from inexpensive, Earth-abundant, starting materials. This transformation occurs in a single stage reactor operating at 300-320 °C and 160-220 bar. The reducing equivalents for these transformations are derived by the reforming of MeOH (to H2 and CO), which thereby serves as a "liquid syngas" in the present case. Water generated by deoxygenation processes is quickly removed by the water gas shift reaction. The Cu-doped PMO serves multiple purposes, catalyzing substrate hydrogenolysis and hydrogenation as well as the methanol reforming and shift reactions. This one-pot “UCSB process” is quantitative, giving little or no biochar residual.
Provided is an overview of these catalysis studies beginning with reactions of the model compound dihydrobenzofuran that help define the key processes occurring. The initial step is phenyl-ether bond hydrogenolysis; however, this is followed by aromatic ring hydrogenation. The complete catalytic disassembly of the more complex organosolv lignin to monomeric units, largely propyl-cyclohexanol derivatives is then described. Operational indices based on 1H NMR analysis are also presented that facilitate holistic evaluation of these product streams that within several hours consist largely of propyl-cyclohexanol derivatives. Lastly, we describe the application of this methodology with several types of wood (pine sawdust, etc.) and with cellulose fibers. The product distribution, albeit still complex, displays unprecedented selectivity towards the production of aliphatic alcohols and methylated derivatives thereof. These observations clearly indicate that the Cu-doped solid metal oxide catalyst combined with sc-MeOH is capable of breaking down the complex biomass derived substrates to markedly deoxygenated monomeric units with increased hydrogen content. Possible implementations of this promising system on a larger scale are discussed.
One-pot Reduction of 5-hydroxymethylfurfural via Hydrogen Transfer from Supercritical Methanol
2. May 2012
Hansen TS, Barta K, Anastas PT, Ford PC, & Riisager A. Green Chemistry. 2012, 14, 2457-2461.
DOI: 10.1039/C2GC35667H
Abstract
Catalytic conversion of HMF to valuable chemicals was achieved over a Cu-doped porous metal
One-pot Catalytic Conversion of Cellulose and of Woody Biomass Solids to Liquid Fuels
1. Aug 2011
Maton TD, Barta K, Iretskii AV, & Ford PC. J. Am. Chem. Soc. 2011, 133, 14090-14097.
DOI: 10.1021/ja205436c
 Abstract
Abstract
Efficient methodologies for converting biomass solids to liquid fuels have the potential to reduce dependence on imported petroleum while easing the atmospheric carbon dioxide burden. Here, we report quantitative catalytic conversions of wood and cellulosic solids to liquid and gaseous products in a single stage reactor operating at 300–320 °C and 160–220 bar. Little or no char is formed during this process. The reaction medium is supercritical methanol (sc-MeOH) and the catalyst, a copper-doped porous metal oxide, is composed of earth-abundant materials. The major liquid product is a mixture of C2–C6 aliphatic alcohols and methylated derivatives thereof that are, in principle, suitable for applications as liquid fuels.
Biomass and sustainable chemistry presentations since 2014
Invited talk: C. Bernt, "Understanding and enhancing th eselectivity of reductive lignin disassembly over doped porous metal oxides"
Christopher M. Bernt, Giovanni Bottari, Jacob Barrett, Megan Chui, Katalin Barta, Susannah L. Scott, Peter C. Ford
250th ACS National Meeting, Boston, MA, August 2015.
Invited talk: P. C. Ford, “Upgrading Porous Metal Oxide Catalysts for the Reductive Disassembly of Lignocellulose to Value Added Platform Chemicals”
Peter C. Ford, Christopher M. Bernt, Megan Chui, Zachary Jones, Giovanni Bottari, Felix Brunner, Hussaya Maneesuwan, Katalin Barta, Alexei Iretski, Susannah L. Scott
249th ACS National Meeting, Novel Catalytic Materials for Renewable Fuels/Chemicals. Denver CO, March 2015
Invited talk: P. C. Ford, “Studies of the reductive disassembly of lignin and lignocellulose using Earth-abundant catalysts”
Peter C. Ford, Katalin Barta, Alexei V. Iretskii, Susannah L. Scott
248th ACS National Meeting, Catalysis Division Symposium: Catalysis for Biomass Conversion. San Francisco CA, August 2014
Invited talk: P. C. Ford, “Reductive disassembly of lignin and lignocellulose using Earth-abundant catalysts”
Peter C. Ford, Katalin Barta, Alexei V. Iretskii, Susannah L. Scott
18th Annual Green Chemistry & Engineering Conference of the ACS, Bethesda MD. June 2014
Contributed Talk: Z. Jones, “Supported metal catalysts for lignin hydrogenolysis”
Zachary Jones, Jessica Wu, Chris Bernt, Bryan Goldsmith, Susannah Scott, Baron Peters, Peter Ford
Symposium on Novel Catalytic Materials for Renewable Fuels/Chemicals, 249th ACS National Meeting, Denver
Contributed Talk: Z. Jones, “Reactions of Lignin Model Compounds over a Hydrotalcite-Derived Copper Catalyst”
Zachary Jones, Jessica Wu, Chris Bernt, Bryan Goldsmith, Susannah Scott, Baron Peters, Peter C. Ford
Symposium on Catalytic Processing of Fossil and Biorenewable Feedstocks, AIChE Annual Meeting, Atlanta GA, 11/19/14
Contributed Talk: A. E. Leak, “The Role of Research Centers in Graduate Students’ Professional Development”
Anne E. Leak, Jason Tillman, Christopher Bernt, Clifford Kubiak, Lubella A. Lenaburg, and Peter C. Ford.
248th ACS National Meeting, Div. Chem Ed. Symp., San Francisco CA, August 2014
Poster: Megan Chui, Breanna Johnson, Christopher M. Bernt, Peter C. Ford, Petra Van Koppen, Darby L. Feldwinn, “Partnership for elementary school outreach: Developing an inquiry-based module to investigate CO2 production, consumption and levels of CO2 in the environment"
248th ACS National Meeting, Div. Chem Ed., San Francisco CA, August 2014
Poster: Zachary Jones, Chris Bernt, Giovanni Bottari, Hussaya Maneesuwan, Susannah Scott, Peter Ford, Katalin Barta, “Probing the Mechanism and Structure of a Lignin Reducing Cu Doped Porous Metal Oxide Catalyst”
248th ACS National Meeting, Div. Inorg. Chem. San Francisco CA, August 2014
Poster: Megan Chui, Danielle Hamann, Anthony T. Tran, Felix M. Brunner, Alexei V. Iretskii, Mauricio Boscolo, Susannah L. Scott, Peter C. Ford, “Tuning the reductive disassembly of lignocellulosic biomass and model compounds using doped porous metal oxides"
248th ACS National Meeting, Div. Inorg. Chem., San Francisco, August 2014
Photochemistry and Small Molecule Bioregulator Presentations since 2014
Invited talk: P.C. Ford, PacificChem, Honolulu, Hawaii December 2015
Invited talk: P.C. Ford, "Strategies for the photochemical uncaging of bioactive small molecules"
10th International Conference on Pharmaceutical Science. Ribeirao Preto, Brazil. September 2015.
Contributed talk: A.W. DeMartino, "Progress towards controlled biological release of CS2, a likely small molecule bioregulator"
Southern California Inorganic Photochemistry (SCIP) Conference, September 12, 2015.
Contributed talk: P. Huang, "Cell-mediated delivery for Photo-activated NO Releasing Moieties"
Southern California Inorganic Photochemistry (SCIP) Conference, September 12, 2015.
Contributed talk: Z. Li, "Rhenium-manganese dinuclear metal carbonyl complexes as interesting photoCORMs"
Southern California Inorganic Photochemistry (SCIP) Conference, September 12, 2015.
Invited talk: P.C. Ford, "Nanoparticle strategies for photochemical delivery of bioactive small molecules"
Gordon Research Conference on Photochemistry, Easton, MA, July 2015.
Invited talk: P.C. Ford, "Nanoparticle strategies for photochemical delivery of bioactive small molecules"
Aregentina National Meeting on Physical and Inorganic Chemistry (aaiFQ). Buenos Aires, April 2015.
Invited talk: P.C. Ford, "Strategies for the photochemical uncaging of bioactive small moleclules"
249th ACS National Meeting, Denver, CO, March 2015
Contributed talk: A.W. DeMartino, "Carbon disulfide: a potential small molecule bioregulator."
Southern California Inorganic Photochemistry (SCIP) Conference, September 27, 2014.
Invited talk: P.C. Ford, "Mechanistic studies of NO and NOx reactions with heme and non-heme iron"
248th ACS National Meeting, San Francisco, CA, August 2014
Poster: Anthony W. DeMartino, Elizabeth S. Levy, Peter C. Ford, "Mechanistic insights and new systems based on 1,1-dithiooxalate, a photochemical precursor to carbon disulfide, a small molecule with biological potential"
248th ACS National Meeting, San Francisco, CA, August 2014
Poster: Meredith A. Crisalli, Lilian P. Franco, Agustin E. Pierri, Po-Ju Huang, Roberto S. da Silva, Peter C. Ford, "Release of nitric oxide from ruthenium centered photoNORMs an their use as small molecule delivery platforms"
248th ACS National Meeting, San Francisco, CA, August 2014